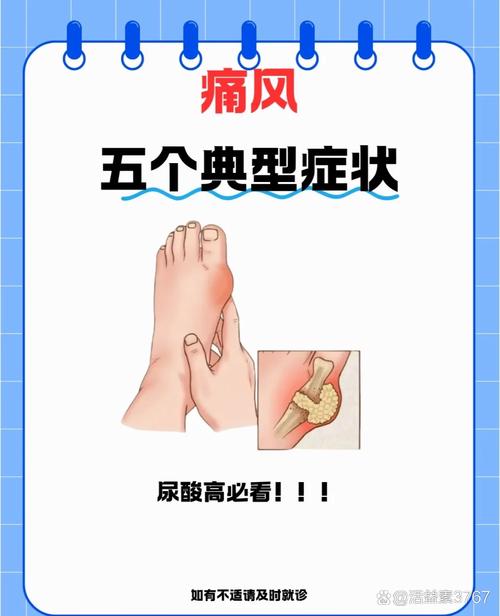
痛风症产生的生化机理主要与体内嘌呤代谢紊乱及尿酸排泄障碍导致的高尿酸血症直接相关,其核心环节是尿酸盐结晶在关节及组织中沉积引发的炎症反应,具体生化过程可分为嘌呤代谢失衡、尿酸生成过多、排泄减少、结晶沉积及炎症级联反应五个阶段。
嘌呤是人体内核酸代谢的终末产物,分为内源性(约占80%,由自身细胞分解产生)和外源性(约占20%,来自高嘌呤食物如海鲜、动物内脏),正常情况下,嘌呤经黄嘌呤氧化酶作用最终生成尿酸,后者通过肾脏排泄(约占70%)和肠道排泄(约占30%)维持动态平衡,当嘌呤代谢关键酶活性异常或尿酸转运功能障碍时,尿酸生成与排泄失衡,导致血尿酸水平超过饱和度(男性>420μmol/L,女性>360μmol/L),形成高尿酸血症。
尿酸生成过多的机制包括:①内源性嘌呤代谢加速:如磷酸核糖焦磷酸合成酶(PRPP)活性过高,或次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HGPRT)缺乏(见于Lesch-Nyhan综合征),导致嘌呤合成增加;②外源性嘌呤摄入过多:高嘌呤食物代谢后直接提升尿酸前体;③细胞大量分解:如肿瘤化疗、溶血性疾病等导致核酸分解加速,产生过量尿酸,尿酸排泄减少则主要与肾脏功能障碍相关:①肾小管分泌减少:尿酸盐转运蛋白(如URAT1、GLUT9)表达异常或功能受抑,导致尿酸重吸收增加;②肾小球滤过率下降:慢性肾病等疾病削弱尿酸滤过能力;③药物影响:如利尿剂、小剂量阿司匹林等竞争性抑制尿酸排泄。
当血尿酸过饱和,尿酸盐结晶析出并沉积于关节软骨、滑膜、肾脏及皮下组织,被巨噬细胞识别为“异物”后,通过Toll样受体(TLR)等模式识别受体激活NLRP3炎症小体,活化的NLRP3炎症小体招募ASC和procaspase-1,切割pro-IL-1β和pro-IL-18为成熟的IL-1β和IL-18,后者诱导中性粒细胞、巨噬细胞浸润,释放TNF-α、IL-6等促炎因子,引发关节红肿热痛等急性炎症反应,尿酸盐结晶还可激活补体系统,产生C5a等趋化因子,进一步放大炎症级联反应,形成痛风急性发作的病理基础,长期结晶沉积可诱发肉芽肿形成,导致慢性痛风石性关节炎和肾功能损害。
以下是相关问答FAQs:

Q1:高尿酸血症一定会发展为痛风吗?
A1:不一定,约5%-12%的高尿酸血症患者会发展为痛风,其余人群可能终身无症状,是否发病取决于血尿酸水平、持续时间、尿酸盐结晶沉积部位及个体免疫反应敏感性,长期血尿酸>540μmol/L者,痛风风险显著升高,需通过控制饮食、药物干预降低发病风险。
Q2:痛风发作时为何血尿酸水平可能正常?
A2:痛风急性发作期,尿酸盐结晶从血液中析出沉积于关节,可能导致血尿酸暂时性下降,造成“正常假象”,部分患者因应激反应导致尿酸排泄短暂增加,或存在检测误差,此时不能仅凭血尿酸结果排除痛风,需结合关节液穿刺(发现尿酸盐结晶)或影像学检查(如双能CT)确诊。